原田 彰宏
大阪大学 医学系研究科 細胞生物学教室
概要・原理
装置・器具・試薬
詳細
Q:なぜマウスを使うの?(細胞ではなくマウスを使う必要性)
A:1)ある分子が必ずしもその細胞(やその細胞が由来する組織)で有用でないかもしれないし、発生のある時期だけに重要かもしれない 2)RNAi自体に問題があることもある(off target effectとか)
Q:マウスを扱ったことがないけれど、使うことは出来る?
A:1)動物倫理をよく理解し、適切な動物実験を行う訓練を受けていれば大丈夫 2)動物を使うのは、そこから組織を取って調べる目的が主なので、形態学の基本(固定、切片の作製法など)は身につけたほうがよい。もし知らなければ、形態が出来る研究室に教えてもらう、ないしは共同研究するなどの手がある。
Q:マウスを使うのにはお金がかかると聞いたけれど、どの位かかるの?
A:かかる費用は下記のように分けられる
1)マウスを作製しない場合
・飼育費
・解析の費用:やることによる
*形態中心だと染色液、パラフィン、エポンなどはそれ程高くないし、免疫染色をするのなら抗体の費用による
・マウスをもらうための費用
:アメリカJackson研究所で色々なマウスを扱っているが、これらは1匹で6〜9万円ほど(輸送料込み)
理研BRCや熊本大学のCARDからも入手可能(連絡先は後述)で、これらからもらう場合は輸送料と微生物検査料などで10〜20万円(ただ共同研究扱いになることもあるので注意)
・飼育費:大学による
例)大阪大学、群馬大学では1日1ケージ(2〜3匹用)で20円強
1系統(遺伝子)を解析するには40〜50ケージ必要→年間30〜40万円
2)マウスを作製する場合
この金額に加え、
・トランスジェニックマウスの場合
ベクター作製は自分でやるとして、そのベクターを送って外注して1 constructで50万円位(筑波大学、熊本大学)
*大学内の施設で受注している場合は多分もっと安い
・ノックアウトマウスの場合
ベクター作製は自分でやるとして、そのベクターを送ってES細胞のスクリーニングをやってもらう:100〜200万円強(筑波大学:人を派遣するか否かによって異なる)
*私の研究室も立ち上がり次第、スクリーニングを共同研究、実費程度で行う予定
相同組み換えをおこしたES細胞を胚盤胞(Blastocyst)に注入してキメラを作ってもらう:50〜100万くらい(筑波大学、熊本大学)
輸送費、マウスの微生物検査などを合計すると200万円以上はかかる
*更にベクター作製の費用と人手はかかる:筑波大学で受託している(50〜100万円以上)
3)gene trap mouseを購入する場合
IGTC(International Gene Trap Consortium)から入手
conditional knockoutにする必要がなければそれらを使うことも可能。まだ少ないが下記のKOMPよりは多い
conditional knockout project (KOMP, EUCOMM)から入手
ただ、全ての遺伝子についてgene trap lineやknockout lineがあるわけではない(というかまだまだ少ない)ので、無ければ自分で作らなければならない。
gene trap mouseを入手
最近Sleeping Beauty (SB)やPiggy Bac (PB)などのtransposonを用いてgene trap mouseが効率よく産出されるようになってきた。しかし共通のリソースや商業ベースでこれらを入手できるようにはまだなっていない。
IGTCについて
数年前からretrovirusなどを感染させて遺伝子を破壊したES細胞がIGTC (International Gene Trap Consortium)から入手できるようになったので、conditional knockoutにする必要がなければそれらを使うことも可能。その場合、ES細胞で送ってもらい(1系統あたり約10万円)、それを日本でinjectionする方法と、向こうでマウスにしてもらう方法(+50万円)がある。
どちらもgermline transmissionすることは確認済み。また、最近conditional knockout projectも進行中なので、そこからES細胞をもらってinjectionしてもらうことも可能(KOMP, EUCOMMから入手可能)。
IGTC(International Gene Trap Consortium)
Knockout Mouse Project (KOMP) Repository
Q:自分でノックアウトマウスを作製する場合、どうやって作ればよいの?
A:一般にはノックアウトマウスの作製において、人為的にはどうしようもないと考えられている以下の2つの技術的な壁があります。
1)homologous recombinationを起こしたES細胞がとれるかどうか
2)germlineにいくかどうか
しかしこれらはかなりの確率で乗り越えることが可能です。そしてそれは作製を始める段階で大体決まってしまいます。 homologous recombinationを起こしたES細胞がとれるかどうかについて
A)long armの長さ
B)negative selection(tkないしはDT-A)を行うかどうか
が大事といわれていますが、それ以上に重要なのは
positive selection markerに何を使うか?
ということであり、我々はpositive selection markerの選択がよいため、homologous recombinantが得られていると考えています。
我々は、まずknockoutする遺伝子がES細胞で発現しているかどうかをRT-PCR法で調べることにしており、ES細胞で発現している分子、発現していない分子に分けて方法を変えています。
A)ESで発現している遺伝子のknockoutではpromoter trap法を用いる
つまり、SA (splice acceptor)-IRES (internal ribosomal entry site)の後ろにpromoterなしのbeta-geo (LacZとneoの融合遺伝子)+poly A signalを付けている
SA-IRESの後ろにbeta-geoを付けた時は、neoを付けた時よりもhomologous recombinationの効率が4ー10倍上がる。
B)ESで発現していない遺伝子ではどうするか?
通常のpromoter付きのbeta-geoを用いるが、その際、beta-geoのcoding regionにmutationが入って、G418耐性が低下しているものを選ぶ(最近使われているものは殆どmutationが直されてしまっているので、古くから使われているものをもらう:当方からも譲渡可能)
neo (mutation+)とbeta-geo (mutation+)の比較をするとbeta-geoの方がhomologous recombinationの効率は高い。
例)synapsin I knockout (Journal of Cell Biology 131, 1789 1995)ではbeta-geoのhomologous recombinationの効率はneoの14倍!
2)germlineにいくかどうか
これは、ES細胞の種類を問わず、質の良いES細胞を用いることに尽きる。
ただ、我々が特に注意しているのは、「germline transmissionが確認されている」とされるES細胞でもcultureのprotocolが譲渡先と自分達の間で微妙に違ったり、入手するまでに劣化する場合もあるため、 targeting vectorを導入する前の普通に培養しているES細胞がgermlineに通るか、必ず事前に確認している。
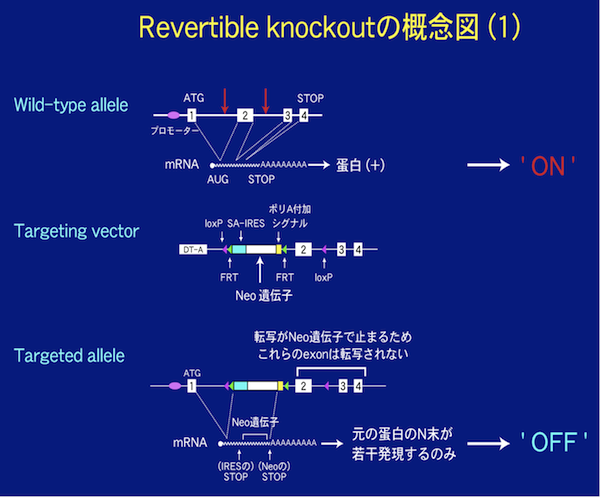
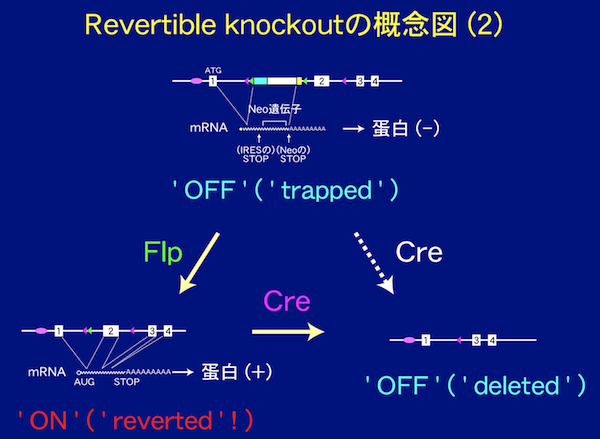
3)表現型を野生型に戻せるgene targeting(revertible gene targeting)について
現在我々はconditional gene targetingについても改善を図っており、表現型を野生型に戻せるgene targeting (‘revertible gene targeting’)を次に紹介します。この手法では
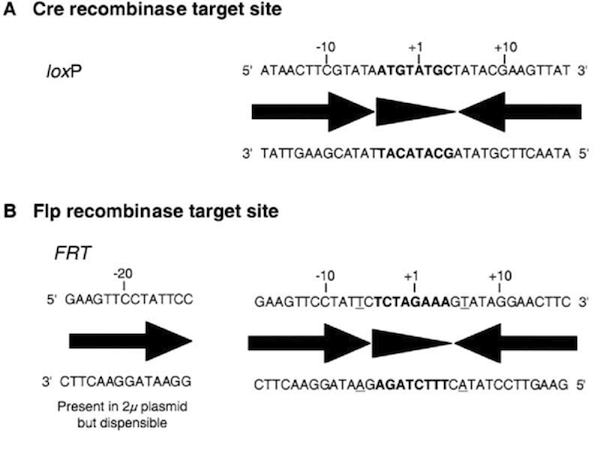
Cre (recombinase) – loxP (target site)
Flp (recombinase) –FRT (target site)
の2種類のrecombinaseとtarget siteの組み合せを用います。(Branda et al. Dev. Cell 6, 7- (2004))
この手法は下記の2つの大きな利点がある。
1)1つのベクターで全身のノックアウトと組織特異的ノックアウトマウスが作製できる。
2)遺伝子の機能回復(rescue)が容易に出来るため、ある遺伝子のノックアウトマウスの表現型がその遺伝子によるものか否かが容易に確認できる(ES細胞はしばしば染色体異常などをおこすため、表現型が本当はその遺伝子によらないこともある)。
Q:ベクターの作製はどうやってやるの?
A:我々はBACをCHORI (Children’s Hospital Oakland Research Institute:URLは最後に掲載)から入手してそこからgenome断片を切り出して使っている。 最近、多くの人はPCRを使って断片を得ているようなので、多分それで出来ると思われる(我々も最近PCRで作製中)。Mutationさえ入らなければ大丈夫と思われるので、近くで作っている人にやり方を聞いてみてください。
Q:マウスは出来たけど、どうやって解析すれば良いの?
A:目標とする組織や細胞がある場合は、そのようなことを考えずに済むので、ここでは触れない。
ただ、多くの場合予想しない表現型を呈するので、上のように考えていても思うようにいかないことも多い。
ではどうするか?
胎生致死の場合とそうでない場合を分けて考える。
胎生致死の場合
Conditional knockoutを作っていなければ、解析しながらでも作り始めるほうが結局時間の節約になる。
解析については、どこまでembryoが生存するか、メスの子宮を開いて調べる。
胎生9,5日以降で死ぬなら、割とmaterialが取れるので解析できることも多いが、それ以前に死ぬと解析が難しくなるので、発生の過程を見たいというのでなければconditionalで解析するのが賢明。
生まれてくる場合
まず、解剖してみてどの臓器が外見上異常か、調べる。
*体の全身の臓器を取り出してから固定、ないしは全身を還流固定して臓器を取り出してパラフィン切片をつくり、HE染色で観察する。それで異常を示す臓器にターゲットを絞って解析する。
しかし、重要な遺伝子と考えていても、実際にノックアウトマウスが生まれると全く異常が外見上から分からず、成長や生殖能にも異常を来さないことも結構ある(どんなにその遺伝子が重要と言われていても、経験上半分くらいはそのようなケースがある)。
Q:全く異常が分からない場合、どうすれば良いの?
A:まず機能の重複を考える。我々はある遺伝子がファミリーを形成する場合は出来るだけそのファミリーの遺伝子の欠損マウスを同時に作製するようにしている 。作製するのが面倒なら、先に述べた入手先で扱っていればそこからもらう。
Q:それでも異常がない場合(または類似の遺伝子が無い場合や類似遺伝子がありすぎて2重欠損マウスが作れない場合)、どうすれば良いの?
A:これについては、その遺伝子の機能を考えながら負荷をかけてみたり(例えば糖代謝に関わる可能性がある遺伝子なら糖負荷試験などをやってみる)、機能試験(長期記憶など)を行ってみる。 またmicroarrayでノックアウトとコントロールでどのような遺伝子の発現が異なるかであたりを付けることも出来る。



